iGenomes
This page is a set of instructions to utilize the Genome parameter of the rnaseq nf-core pipeline. It requires you to download the necessary resources.
iGenomes are a collection of sequence and annotation files for commonly analyzed genomes. Each iGenome contains data for one species, downloaded from one source (UCSC, NCBI, or Ensembl), for one genomic build. They are available and easy to use, but not recommended because gene annotations are out of date and some iGenomes references (e.g., GRCh38) point to annotation files that use gene symbols as the primary identifier.
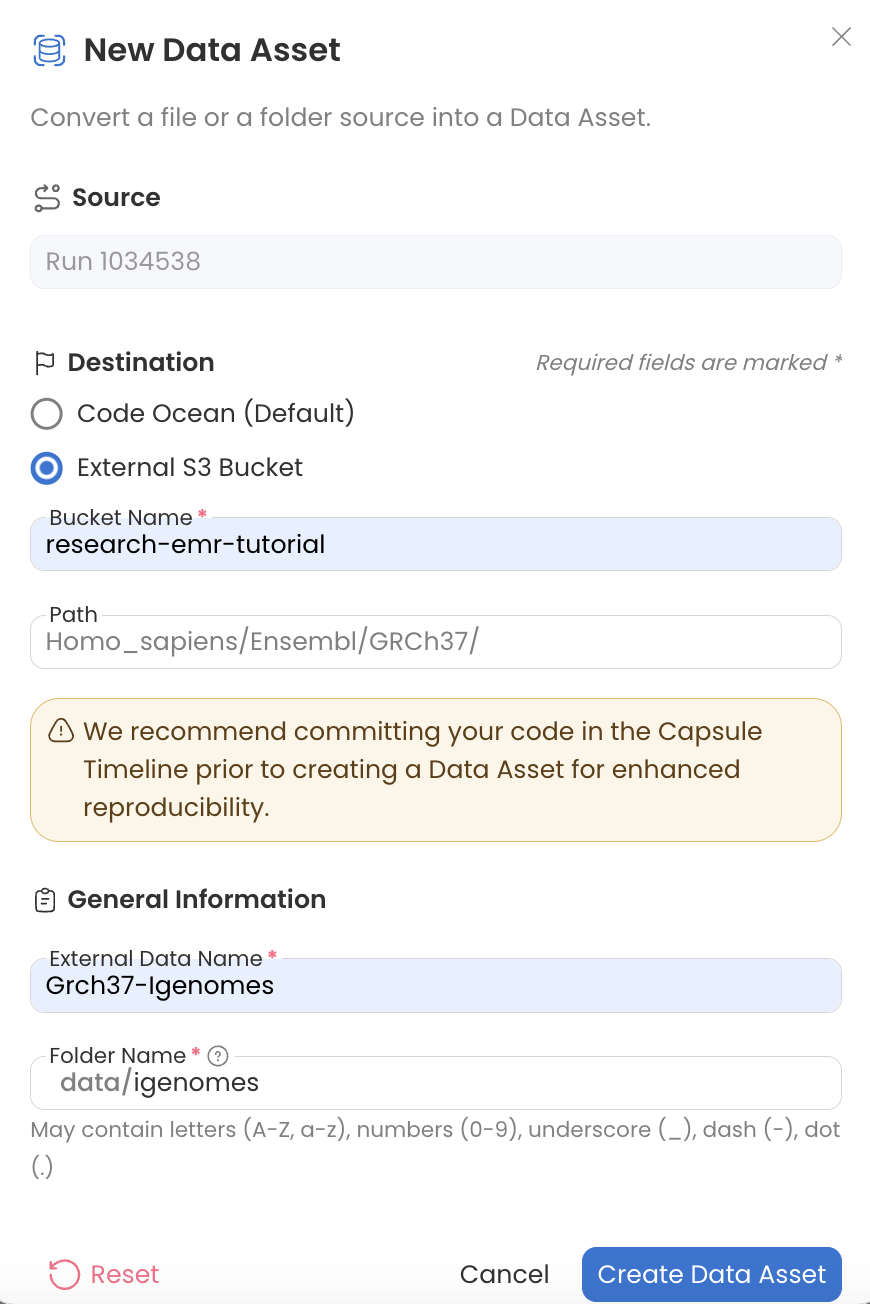
Source External Data
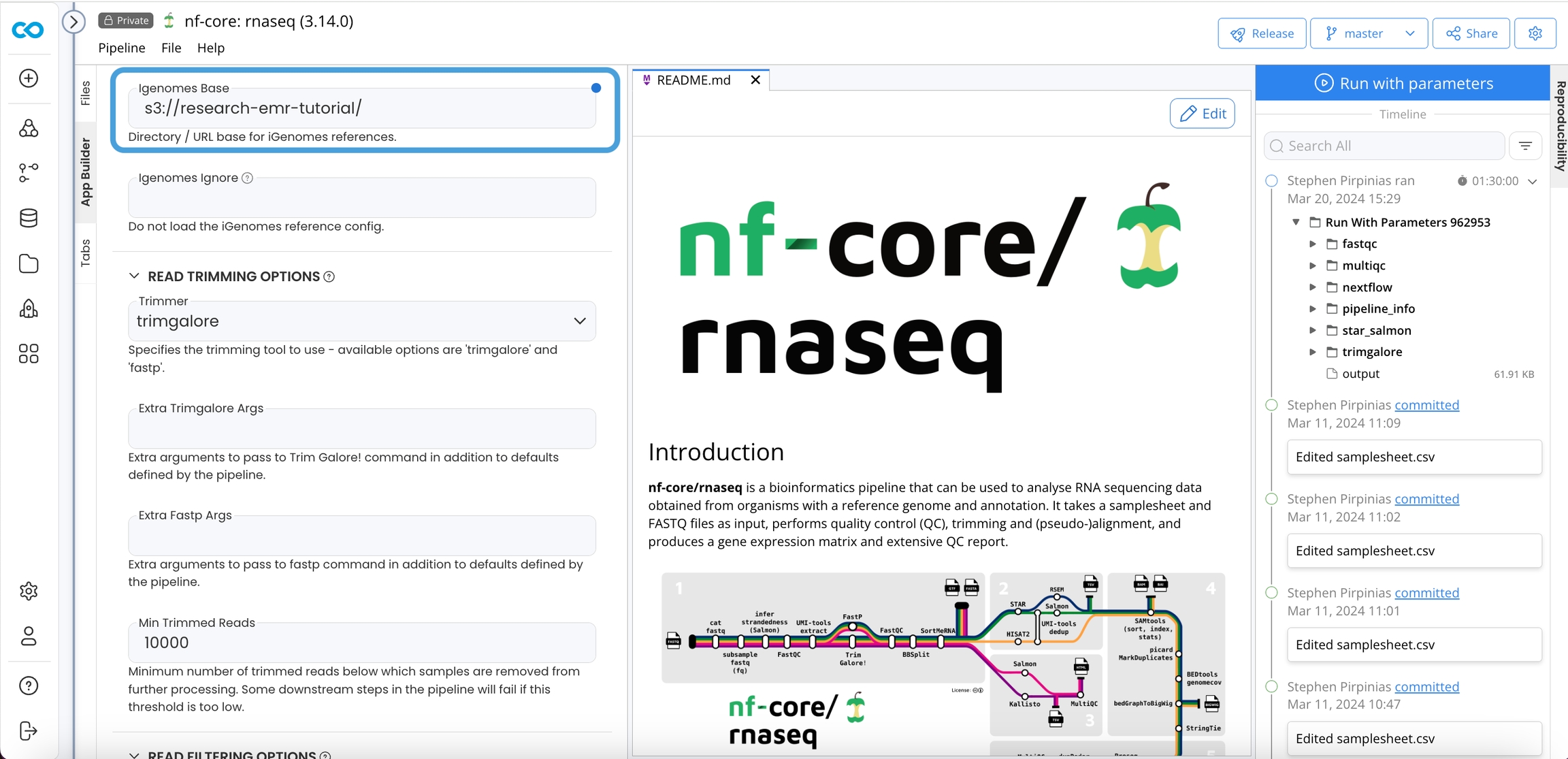
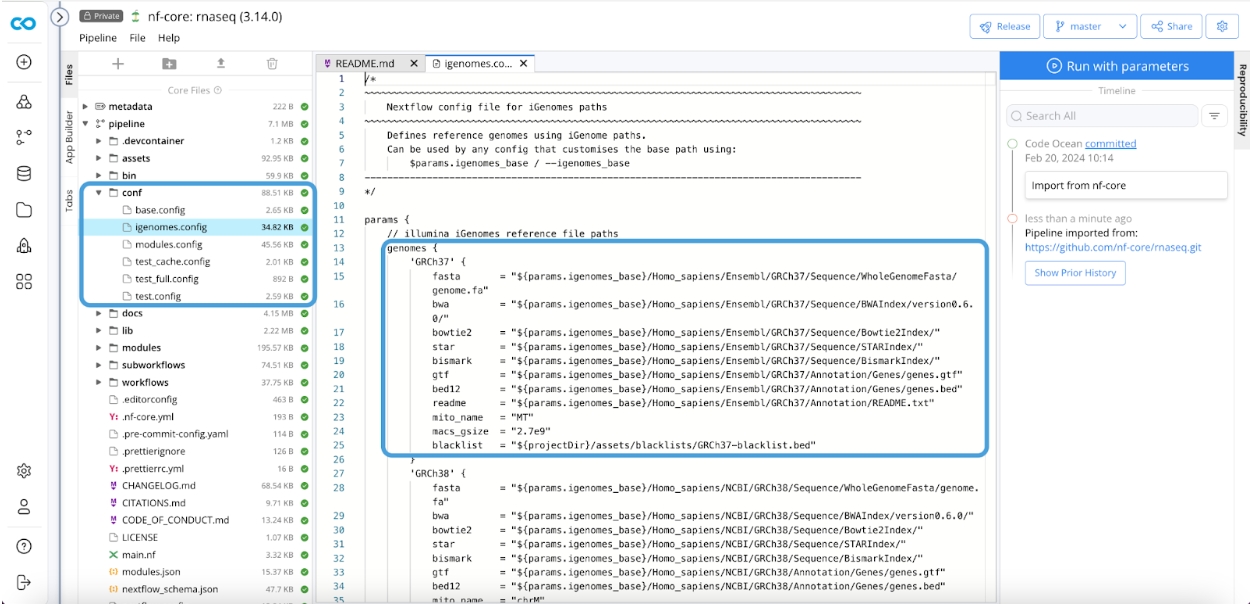
nf-core requires a configuration file to use iGenomes resources. That file is pipeline/conf/igenomes.config.
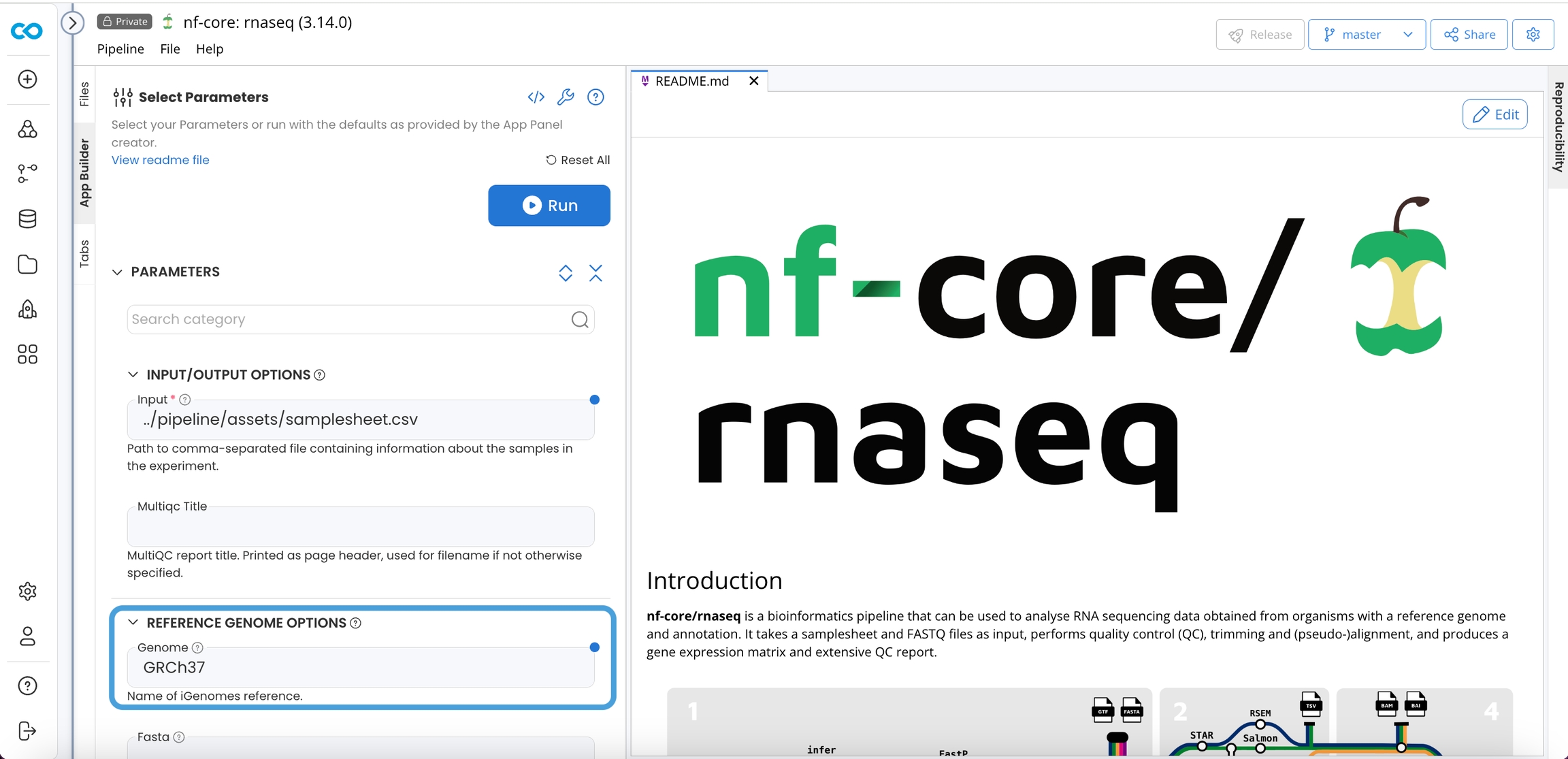
When you set the Genome parameter to any of the choices in the igenomes.config, the variables described in the json will be assumed. It is critical your S3 directory structure match this file and path structure because your igenomes.base will be the prefix of your S3 bucket the contents reside within. That is the text in {curly brackets}.
Downloading iGenomes
You can use this service to create a query to download the required files. For instance,
This command can be run from a Capsule by Reproducible Run or Cloud Workstation. It will download the necessary resources required for the GRCh37 into your Results folder.
Next create an External Data Asset from the Results.
Using an External Data Asset allows you to leave the Igenomes base App Panel parameter and igenomes.config file as is. Using an Internal Data Asset would require you to edit these.
To use External Data Assets in a Pipeline, Assumable Roles must be configured by a Code Ocean admin.
The path structure must be identical to the json (picture above) for parameter - GRCh37.
If your directory structure is as shown in the iGenomes configuration file, iGenomes resources for your chosen genome will be downloaded into your bucket.